For decades, cancer metabolism was defined by the Warburg effect. However, new technologies are revealing a far more flexible metabolic strategy in which tumors dynamically shift between fuels to survive environmental and therapeutic pressures.
T-cells of the immune system attacking growing cancer cells
Rethinking the metabolism of cancer
For decades, cancer metabolism appeared to follow a relatively simple rule. Tumor cells preferentially convert glucose to lactate even when oxygen is available, a phenomenon known as the Warburg effect. First described in the early twentieth century, this metabolic behavior became one of the earliest biochemical hallmarks of cancer that shaped scientific thinking about tumor metabolism for generations.1
The Warburg effect suggested that cancer cells rely heavily on glycolysis, a less efficient pathway for energy production, even under conditions where mitochondrial respiration would normally dominate. This observation helped establish the idea that cancer metabolism is fundamentally distinct from that of healthy tissues.2 This is why the search for metabolic vulnerabilities in cancer has focused largely on altered glucose metabolism.
Recent advances, however, are revealing a more complex picture. Technologies such as metabolic tracing, high-resolution metabolomics, and spatial profiling now allow researchers to examine how tumors process nutrients directly within living tissues.3 These approaches suggest that many cancers do not rely on a single metabolic strategy. Instead, tumor cells appear capable of dynamically shifting between different energy pathways depending on oxygen availability, nutrient supply, and therapeutic stress.4
Cancer cells can exploit a wide range of metabolic fuels, including glucose, glutamine, fatty acids, and lactate produced within the tumor microenvironment.5 This capacity for metabolic adaptation, often referred to as metabolic plasticity, suggests that tumor metabolism is not fixed but highly flexible.6 Understanding how cancer cells reorganize their energy systems may therefore become essential for explaining tumor growth and therapeutic resistance.
Cancer’s expanding menu of fuels
While the Warburg effect highlighted the importance of glucose metabolism in cancer, subsequent research has shown that tumor cells frequently rely on additional fuels to sustain growth and survival. Rather than operating through a single dominant pathway, cancer metabolism appears to function as a flexible network that can draw on multiple nutrient sources.7
Among the most prominent of these alternative fuels is glutamine, an abundant amino acid that supports a variety of biosynthetic processes. Rapidly dividing tumor cells use glutamine as both a carbon and nitrogen source, enabling the production of nucleotides, amino acids, and other cellular building blocks.8 Many cancers rely heavily on glutamine metabolism to support biosynthesis, redox balance, and cellular proliferation.9
Tumor cells can also exploit lipid metabolism as an alternative energy strategy.10 Fatty acids may be broken down through β-oxidation to generate ATP and metabolic intermediates, particularly in solid tumors growing under nutrient-limited conditions. Some tumors even rely on external lipid sources supplied by surrounding tissues or circulating lipoproteins, illustrating how cancer cells can draw metabolic support from their environment.11
Perhaps most surprising is the growing recognition that metabolic byproducts produced by one population of tumor cells can serve as fuel for another. Lactate, once considered merely a waste product of glycolysis, is now known to function as a valuable metabolic substrate in certain cancers.12 Metabolic tracing studies in human lung tumors have demonstrated that lactate can be taken up and oxidized by tumor cells, contributing carbon to the tricarboxylic acid (TCA) cycle.13
Diverse metabolic fuels that tumor cells use are not determined by cellular metabolism alone. The surrounding environment within the tumor also plays a decisive role in shaping metabolic behavior.14
A metabolic landscape inside tumors
Unlike healthy tissues with stable blood supply, tumors frequently develop in environments where oxygen and nutrients are unevenly distributed. As tumors grow rapidly, their expanding mass can outpace the development of new blood vessels, creating regions of hypoxia and nutrient limitation. These environmental constraints strongly influence how cancer cells generate energy and sustain growth.15
In oxygen-poor regions of tumors, cells often rely more heavily on glycolysis, converting glucose into lactate to sustain ATP production despite limited mitochondrial respiration. In contrast, tumor cells located closer to blood vessels may have greater access to oxygen and nutrients, allowing them to utilize oxidative phosphorylation more efficiently. This spatial variation creates metabolic diversity within tumors, with neighboring cells operating under very different metabolic conditions.16
Research has also revealed that tumor cells can engage in forms of metabolic cooperation. Experimental studies have shown that lactate produced by glycolytic tumor or stromal cells can be taken up and used for oxidative metabolism by more oxygenated tumor cells.17
Such interactions illustrate how tumor metabolism can function as a complex ecosystem rather than a collection of clonally identical cells.
The tumor microenvironment also includes immune cells, stromal cells, and other nonmalignant components that compete with cancer cells for available nutrients.18 Studies of the tumor microenvironment have shown that different cell types, including tumor cells and immune cells, compete for key nutrients and display distinct metabolic programs.19 As a result, metabolism has emerged not only as a property of cancer cells themselves but also as a defining feature of the broader tumor ecosystem.
In addition to environmental pressures within tumors, cancer therapies themselves can impose metabolic stress that forces tumor cells to adapt.20
Metabolism as a survival strategy
Cancer therapies disrupt cellular processes that are tightly linked to metabolism, including growth signaling, DNA replication, and redox homeostasis. In response, tumor cells may reorganize their metabolic pathways in order to maintain energy production and survive treatment.21
One example of this adaptation has been observed in tumors exposed to targeted therapies that inhibit oncogenic signaling pathways. Experimental studies have shown that pancreatic cancer cells that survive oncogene inhibition can become dependent on mitochondrial oxidative phosphorylation for continued growth.22
Chemotherapy and other cytotoxic treatments can also trigger metabolic changes. Because many anticancer drugs generate oxidative stress or damage cellular components, surviving tumor cells may enhance metabolic pathways that support antioxidant defenses and cellular repair.23 Increasing evidence suggests that metabolic plasticity may therefore represent an important mechanism by which tumors evade therapeutic pressure. Rather than being locked into a single metabolic state, cancer cells appear capable of reorganizing their metabolic networks to accommodate the stresses imposed by treatment.24
Understanding these dynamic metabolic responses has become possible largely due to recent technological advances that enable researchers to observe tumor metabolism with unprecedented detail.
New tools for watching tumor metabolism
Much of the growing interest in metabolic plasticity has been driven by technological advances that now allow researchers to study tumor metabolism with far greater precision than was previously possible.25
For many years, investigations of cancer metabolism relied largely on experiments performed in cultured cells, which often fail to capture the complex conditions present in living tumors.26
One important advance has been the use of stable isotope tracing to track how tumors metabolize specific nutrients in vivo. By introducing stable isotope–labeled nutrients such as glucose or glutamine, researchers can trace the movement of carbon atoms through metabolic pathways and identify which metabolic pathways are actively used by tumor cells.27
Technologies such as single-cell sequencing and spatial transcriptomics are also allowing scientists to map metabolic programs across different regions of tumors. These approaches have demonstrated that tumor metabolism can vary widely even within a single tumor mass, reflecting differences in oxygen levels, nutrient availability, and interactions with surrounding cells.28
Advances in high-resolution metabolomics are further expanding the ability to analyze metabolic networks at scale. Using sophisticated mass spectrometry techniques, researchers can now measure hundreds of metabolites simultaneously, providing a systems-level view of metabolic activity in cancer cells and their surrounding environments.29
Targeting a moving metabolic target
The growing appreciation of metabolic plasticity is beginning to reshape how researchers think about targeting metabolism in cancer.
Over the past decade, several therapeutic strategies have attempted to inhibit individual metabolic pathways, such as glycolysis or glutamine metabolism. While some of these approaches have shown promise in experimental settings, clinical results have often been inconsistent.30
One reason may be the adaptability that characterizes tumor metabolism. If cancer cells can shift between multiple energy pathways, blocking a single metabolic route may simply encourage tumors to rely on alternative fuels.
Recognizing this flexibility has prompted a broader shift toward studying metabolism as an interconnected network rather than a collection of isolated pathways.
Researchers are increasingly exploring how combinations of metabolic interventions, or therapies that exploit metabolic dependencies that arise during treatment, might produce more durable responses.31
Although many questions remain, the emerging view of cancer metabolism suggests that tumors are far more adaptable than once believed.
Understanding how cancer cells reorganize their energy systems in response to environmental and therapeutic pressures may ultimately be essential for developing therapies that can keep pace with this remarkable metabolic flexibility.
LObbYING fOR CHANGE
WHEN YOUTH DOESN’T EQUATE SAFETY
COPPAFEEL! AND THE COST OF BEING “TOO YOUNG” FOR BREAST CANCER
BY ANNE JÄKEL
Breast cancer is the most commonly diagnosed can- cer in women worldwide, with approximately 2.3 million new cases in 2022 alone, according to the World Health Organization (WHO).1 In the United King-dom, around 55,000 women are diagnosed each year.2
Survival rates have improved substantially due to earlier detection and advances in systemic therapy. However, early detection strategies are largely age-based. In the UK, routine breast screening is offered to women aged 50 to 71.3
Photo courtesy of CoppaFeel!
While breast cancer is less common in women under 30, it does occur. Younger patients are more likely to be diagnosed at a later stage and may present with biologically more aggressive disease subtypes.4,5
Despite this, young women reporting breast symptoms frequently encounter reassurance based primarily on age. The phrase “you’re too young” continues to appear in patient narratives.
A diagnosis at 23, and a response todiagnostic bias
Kris Hallenga was diagnosed with stage IV breast cancer at the age of 23. Prior to her diagnosis, she had sought medical advice for symptoms but encountered age-related assumptions that delayed further investigation. Her experience led to the founding of CoppaFeel!, a UK-based breast cancer awareness charity established in 2009.6
CoppaFeel! was created to address a specific problem: young people were not routinely encouraged to understand their bodies or seek medical attention promptly for breast changes. The charity focuses on symptom awareness and promotes regular self-checking across all genders and age groups.6
The initiative began as a personal campaign but has evolved into a national organization delivering education programs in schools and universities, collaborating with healthcare professionals, and running nationwide awareness campaigns.6
The structural issue: age-based reassurance
Breast screening programs are designed for population-level risk reduction. Age thresholds are determined by incidence data and cost-effectiveness modeling. However, screening policy does not eliminate the need for symptom-based vigilance at any age. Studies have demonstrated that younger breast cancer patients may experience longer diagnostic intervals, partly because symptoms are less readily attributed to malignancy in this age group, contributing to provider-related delay.7-8
Diagnostic delay has been associated with more advanced disease at pres- entation in population-based studies and is linked to worse outcomes.9,10
CoppaFeel! challenges the cultural assumption that youth equates to safety. The organization argues that awareness and responsiveness should not be age-restricted. Its campaigns emphasize that breast cancer, while rarer in younger women, is not impossible.6
From awareness to advocacy
CoppaFeel! has moved beyond awareness messaging into structured advocacy.
The organization campaigns for: • Improved symptom education within secondary schools and universities • Greater primary care awareness of young-onset breast cancer • Public health campaigns inclusive of younger age groups • Reduced stigma around breast health discussions
The charity works with medical advisors and policymakers while maintaining its youth-focused communication strategy.6
Relevance to oncology practice
Young-onset cancers are receiving increasing attention across tumor types. Emerging data suggest rising incidence trends in early-onset breast cancers in several high-income countries.11 Although causality remains under investigation, the clinical reality is that more young adults are presenting with advanced malignancies. Oncology professionals are positioned to influence this trajectory by: • Supporting education initiatives aimed at primary care • Encouraging symptom-based referrals without age bias • Promoting fertility preservation counseling at diagnosis • Advocating for research focused on early-onset tumor biology Diagnostic bias is rarely intentional.
It arises from probability-based reasoning embedded within clinical training. However, rare does not mean negligible.
The cost of dismissing outliers can be measured in stage migration and survival.
Photo courtesy of CoppaFeel!
What must change
Three areas of reform emerge from the CoppaFeel! model:
1. Awareness and education
Embed cancer awareness earlier, with more “teachable moments” across the healthcare journey so younger people know what to look for and how to check their bodies.
2. Understanding risk and ensuring faster routes to diagnosis
Ensure rising cancer rates in people below screening age are reflected in referral pathways and diagnostic decision-making.
3. Age-inclusive research and innovation
Require research funding, clinical trials and data collection to meaningfully include younger people and capture long-term outcomes.
Young-onset disease may differ molecularly from postmenopausal breast cancer.12-14
A legacy of structural change
Kris Hallenga died in 2024 but CoppaFeel! continues to operate nationally. Its impact illustrates how patient-led initiatives can influence public health dialogue and clinical culture.
The organization’s central message remains clear: awareness is not optional, and age is not immunity.
CoppaFeel! demonstrates how personal diagnosis can evolve into structural advocacy, and how cultural change can precede policy reform.
ONCOLOGY BREAKTHROUGHS
CIRCULAR DNA DRIVERS
EXTRACHROMOSOMAL DNA: A HIDDEN ENGINE OF CANCER EVOLUTION
BY ANĐELIKA KALEZIĆ
Beyond chromosomes lies a hidden layer of the cancer genome— circular DNA elements that amplify oncogenes, accelerate tumor evolution, and may soon reshape cancer diagnostics and therapy.
When the cancer genome escapes the chromosome
Cancer genomes are typically described through the lens of genomic instability, mutations and structural alterations within chromosomes. Yet in many aggressive tumors, some of the most powerful cancer-driving genes are not confined to chromosomes alone. Instead, they reside in multiple amplified copies on extrachromosomal DNA (ecDNA), circular DNA fragments that exist independently within the nucleus.1
Although these structures were first observed decades ago as small chromatin bodies known as “double minutes,” their functional significance has only recently become clear.2 Large genomic analyses now indicate that ecDNA occurs in a substantial fraction of human cancers and is rarely detected in healthy tissues.
Importantly, tumors harboring ecDNA frequently contain amplified oncogenes and show evidence of accelerated genetic evolution.3 This discovery has shifted attention toward ecDNA as a potential driver of tumor progression and therapeutic resistance.
Circular DNA with unusual genetic power
In non-malignant cells, DNA is organized into chromosomes that segregate evenly during cell division. In contrast, extrachromosomal DNA replication follows non-Mendelian inheritance rules.
These circular DNA elements lack centromeres, meaning they do not attach to the mitotic spindle in the same regulated way as chromosomes do. As a consequence, ecDNA is distributed unevenly between daughter cells during cell division.4
This unusual inheritance pattern is a major contributor to intratumoral heterogeneity that allows tumors to generate significant genetic diversity within a single population of cancer cells. Individual cells may acquire different numbers of ecDNA molecules, resulting in variable oncogene copy numbers across the tumor tissue.5
Many ecDNA elements carry amplified oncogenes, including genes such as EGFR, MYC, MDM2, and CDK4, which play central roles in tumor proliferation and survival. Because ecDNA can exist in high copy numbers, it often drives exceptionally strong oncogenic signaling.
In effect, ecDNA functions as a mobile amplification system capable of rapidly adjusting oncogene dosage.6
Accelerating tumor evolution
The evolutionary implications of ecDNA are potentially quite significant. Cancer progression depends on genetic variation and selection.
The random segregation of ecDNA during cell division creates precisely this kind of variation, generating populations of cells with different oncogene copy numbers.7
Recent studies have also shown that ecDNA molecules can cluster together within the nucleus to form transcriptionally active hubs. These clusters bring oncogenes into proximity with regulatory elements that enhance transcription, resulting in high levels of gene expression and effectively expanding the regulatory landscape of transcription in cancer cells.8
Such mechanisms enable tumors to rapidly increase oncogene activity when growth signals are advantageous. Under selective pressure, including targeted therapy, cells carrying favorable ecDNA amplifications may quickly expand. In this way, ecDNA acts as a powerful engine of tumor evolution.9
Clinical consequences: ecDNA and agressive disease
Accumulating evidence suggests that the presence of ecDNA correlates with aggressive tumor behavior. ecDNA has been detected across multiple cancer types, including aggressive glioblastoma, sarcoma and esophageal carcinoma. Tumors harboring ecDNA often display increased genomic instability and higher levels of intratumoral heterogeneity.10
Clinically, these characteristics translate into more challenging disease courses. Several studies have reported that patients whose tumors contain ecDNA amplifications experience poorer outcomes compared with patients lacking these structures.10
The ability of ecDNA to dynamically alter oncogene copy numbers as well as drive enhanced chromatin accessibility and rewire regulation of gene expression may contribute to rapid tumor adaptation and early relapse.11
These observations have generated interest in ecDNA as a prognostic biomarker and potential therapeutic target that may help identify and treat tumors with particularly rapid progression.
Toward ecDNA-based diagnostics
Beyond its prognostic implications, ecDNA may also contribute to new diagnostic strategies. Because ecDNA contains tumor-specific genomic alterations, fragments derived from extrachromosomal DNA can potentially be detected in circulating tumor DNA.12
Early research suggests that ecDNA signatures in blood samples may help distinguish cancer patients from healthy individuals and may provide information about tumor subtype. In lung cancer, for example, circulating ecDNA profiles have demonstrated promising diagnostic accuracy in preliminary studies.13 Moreover, ecDNA is associated with poor outcomes in multiple cancers including pediatric solid tumors.14
Although this approach remains under active investigation, it raises the possibility that ecDNA analysis could complement existing liquid biopsy technologies.15 By revealing patterns of oncogene amplification and genomic rearrangement, ecDNA detection may provide an additional layer of molecular information about tumor biology.
Monitoring tumor evolution during therapy
Another potential application of ecDNA lies in monitoring treatment response. Because ecDNA copy numbers can change dynamically under therapeutic pressure, tracking these changes may reveal how tumors adapt during treatment.16
For instance, amplification of oncogenes carried on extrachromosomal DNA can change dynamically during tumor progression and in response to targeted therapies that suppress oncogenic signaling pathways. In some cases, tumor cells transiently eliminate ecDNA copies during drug treatment, reducing oncogene expression.
When therapy is withdrawn, ecDNA- bearing clones can reemerge, restoring oncogene amplification and promoting therapeutic resistance.17
These shifts could potentially be detected through serial genomic monitoring in clinical settings. If validated in clinical studies, ecDNA profiling could become a tool for identifying emerging drug resistance before it becomes clinically apparent.18 Such an approach would extend the concept of liquid biopsy from simple mutation detection to the monitoring of structural genome evolution.
Can ecDNA become a therapeutic target?
The discovery of ecDNA also raises the possibility of new therapeutic strategies. Because ecDNA is structurally and functionally distinct from chromosomal DNA, it may depend on unique molecular processes for replication and maintenance potentially making it a pan-cancer therapeutic target.19
Researchers are exploring whether these differences could expose vulnerabilities that can be exploited therapeutically. Strategies under investigation include disrupting the replication mechanisms that sustain ecDNA amplification, interfering with the transcriptional hubs formed by ecDNA clusters, or exploiting the structural instability of ecDNA to selectively damage tumor cells that depend on these elements.20
Although these approaches remain at an early stage, they highlight a broader shift in oncology research.
Rather than focusing solely on individual oncogenic mutations, scientists are increasingly examining the genomic architecture and structural features that allow tumors to evolve and adapt under selective pressure.
A new layer of the cancer genome
The recognition of extrachromosomal DNA adds a new dimension to our understanding of cancer genetics. Rather than viewing the cancer genome as a relatively static collection of chromosomal alterations, it is increasingly clear that tumors can exploit dynamic genetic elements such as extrachromosomal DNA to reshape oncogene copy number and gene expression.
As research progresses, ecDNA may influence several areas of clinical oncology, including cancer diagnostics, biomarker development, and therapeutic innovation. For clinicians and researchers alike, these circular DNA structures represent more than a biological curiosity. They reveal a previously underappreciated mechanism through which tumors generate diversity, adapt to therapy, and evolve over time.
In doing so, ecDNA challenges traditional views of the cancer genome and opens new avenues for understanding, and potentially manipulating, the evolutionary dynamics that drive cancer progression.
ONCOLOGY BREAKTHROUGHS
MULTIPLE MYELOMA
IFM2017-03: DEXAMETHASONE -SPARING DARATUMUMAB – LENALIDOMIDE SETS A NEW BENCHMARK FOR FRAIL, NEWLY DIAGNOSED MYELOMA
BY ANNE JÄKEL
Treating newly diagnosed multiple myeloma (NDMM) in older patients with frailty remains one of the most difficult balancing acts in hematologic oncology: regimens must be potent enough to achieve deep disease control, yet tolerable enough to avoid early discontinuation, functional decline, and infectious morbidity. Steroids are a particular double-edged sword in this population.1 Dexamethasone improves short-term cytoreduction and remains embedded in many frontline backbones, but prolonged exposure can exacerbate infections, metabolic and cardiovascular toxicity, neuropsychiatric effects, and sarcopenia. These are factors that disproportionately destabilize frail patients. Against this backdrop, the phase III IFM2017-03 trial directly tested a clinically pragmatic hypothesis: can we preserve (or even improve) efficacy by intensifying immunotherapy with daratumumab while sharply limiting dexamethasone exposure?1
Published in The Lancet Oncology, IFM2017-03 is notable not only for its results, but also for its intent: it is, to current knowledge, the first phase III trial designed specifically for frail, older patients with NDMM.1
Study design and patient population
IFM2017-03 was a prospective, randomized, open-label, multicentre phase III trial conducted across 61 Intergroupe Francophone du Myélome (IFM) centres in France.
Eligible patients were ≥65 years with NDMM, measurable disease, and frailty defined by an ECOG-proxy frailty score ≥2 (built from age, ECOG performance status, and Charlson Comorbidity Index). Patients were randomized 2:1 to a dexamethasone-sparing regimen of subcutaneous daratumumab (1800 mg) plus lenalidomide (days 1–21 of a 28-day cycle) with dexamethasone used only during the first two cycles (as daratumumab premedication), or to the control regimen of lenalidomide plus weekly dexamethasone continued until progression/toxicity.
The primary endpoint was progression-free survival (PFS), with key secondary endpoints including response depth, minimal residual disease (MRD), overall survival (OS), and tolerability.1
The enrolled cohort reflects real-world geriatric myeloma more closely than many registrational studies. Median age was 81 years (IQR 77–84), and 61% were older than 80. Nearly half had ECOG ≥2, about one-third had ISS stage III disease, and 15% had high-risk cytogenetics.1 This is precisely the population in which clinicians most acutely feel the limitations of standard triplets: toxicity-driven discontinuation and infection risk can erase the theoretical benefit of intensified therapy.
Efficacy results
IFM2017-03 delivered a striking PFS advantage with the dexamethasone-sparing daratumumab–lenalidomide approach. At a median follow-up of 46.3 months, median PFS was 53.4 months (95% CI 35.3–not reached) with daratumumab–lenalidomide and limited dexamethasone, versus 22.5 months (16.5–39.0) with lenalidomide– dexamethasone (HR 0.51, 95% CI 0.37–0.70; p<0.0001).1
In practical terms, the experimental strategy more than doubled median disease control in a cohort in which long remissions are typically difficult to achieve.
Importantly, efficacy was not achieved at the cost of delayed kinetics. Rates of very good partial response (VGPR) or better were higher with the dexamethasone-sparing arm (69% vs 51%), and this separation was already evident early in treatment (cycle 4), suggesting that early steroid discontinuation did not blunt initial cytoreduction when daratumumab was added.
Complete response rates were also substantially higher (34% vs 12%).1 MRD negativity (10% vs 4%) numerically favored the daratumumab–lenalidomide arm but did not reach significance, and missing/unassessable MRD results were common, highlighting the feasibility challenges of marrow-based MRD in very old, frail patients.1
Although PFS was the primary endpoint, the OS signal reinforces clinical relevance. Median OS was not reached in the dexamethasone-sparing group versus 47.3 monthsin the control group (HR 0.52, 95%CI 0.35–0.77), with estimated 4-yearOS rates of 68% versus 48%, respectively.1
In a frail population where competing risks are substantial, a durable OS separation of this magnitude is particularly compelling.
Safety and tolerability
At first glance, toxicity patterns appear paradoxical: grade 3–5 adverse events were more frequent with daratumumab–lenalidomide (89% vs 79%), driven largely by hematologic events, especially neutropenia (55% vs 24%).1
Yet the outcomes that matter most in frail practice, such as serious adverse events, infection severity, and discontinuation, were broadly comparable, and in some clinically meaningful dimensions potentially improved.
Grade 3–5 infections occurred at similar rates between arms (20% vs 21%).1
This is notable because prior data in frail subsets have raised concerns that adding daratumumab to lenalidomide–dexamethasone increases severe infections, particularly pneumonia, in older and vulnerable patients.
The MAIA trial established daratumumab–lenalidomide–dexamethasone (D-Rd) as a frontline standard for transplant-ineligible NDMM, and frailty analyses have suggested higher infection burdens when daratumumab is layered onto steroid-containing backbones.2
IFM2017-03 offers a mechanistically coherent counterpoint: if steroids are a major driver of infectious susceptibility, then minimizing dexamethasone may allow clinicians to capture the immunotherapeutic benefit of anti-CD38 therapy without proportionally amplifying infectious harm.
Treatment discontinuation due to adverse events was numerically lower with the dexamethasone-sparing regimen (18% vs 24%), despite substantially longer exposure (median treatment duration 31.6 vs 14.3 months).1
This matters because discontinuation is not merely an endpoint but a real-world mechanism through which frailty translates into inferior survival. Interestingly, lenalidomide dose intensity was lower in the daratumumab–lenalidomide group, and 14% discontinued lenalidomide while continuing daratumumab monotherapy, underscoring how dose tailoring and simplified maintenance patterns may be integral to making prolonged therapy feasible in the oldest patients.1
Microscopic view of bone marrow slide, showing multiple myeloma
Clinical context and practice implications
IFM2017-03 arrives at a moment when myeloma care is increasingly stratified not only by cytogenetics and MRD, but by biological age, geriatric vulnerability, and the tolerability ceiling of continuous therapy.
The International Myeloma Working Group frailty framework helped formalize frailty as a prognostic and treatment-defining variable, yet practical barriers have limited routine implementation, making simplified tools and trial designs like IFM2017-03 particularly valuable.3
A key interpretive nuance is thechoice of control arm: lenalidomide–dexamethasone, rather than D-Rd. However, the investigators’ rationale is historically consistent: Rd remained widely used at protocol conception and continues to be selected for frail patients in many settings due to oral convenience and concerns about triplet tolerability.
IFM2017-03 therefore answers a question that is both biologically and operationally relevant: for the frail patient in whom clinicians hesitate to deliver continuous steroids, can daratumumab–lenalidomide with early steroid discontinuation deliver superior outcomes without unacceptable toxicity? The data strongly suggest yes.1 For clinicians, the trial supports three practical shifts:
Steroid minimization can be a deliberate strategy, not a reluctant compromise. IFM2017-03 suggests that, when paired with daratumumab, limiting dexamethasone to the initial cycles can preserve rapid response while improving the therapeutic ratio in frail NDMM.1
Hematologic toxicity requires proactive management, but does not inevitably translate into more severe infections. High neutropenia rates emphasize the need for close monitoring, dose adjustment, and supportive care, yet severe infection rates remained comparable, potentially reflecting the benefit of reducing chronic steroid exposure.1
Dose-adaptive lenalidomide “de-escalation” may be the hidden enabler of long treatment duration. The low relative dose intensity reported suggests many frail patients functionally transition to lower-dose maintenance; future protocols may benefit from defining this approach prospectively rather than treating it as reactive toxicity management.1
Conclusion
IFM2017-03 establishes dexamethasone-sparing daratumumab–lenalidomide as a major advance for frail, newly diagnosed myeloma, a population long underserved by conventional trial designs. With median PFS extending beyond four years (53.4 months) and a meaningful OS signal, the regimen offers a compelling balance of efficacy and tolerability despite increased hematologic toxicity.
More broadly, the trial reframes steroid exposure as a modifiable determinant of treatment feasibility in geriatric myeloma. As frontline therapy continues to intensify, IFM2017-03 provides rare phase III evidence that “less steroid” can coincide with “more benefit” when modern immunotherapy is deployed intelligently.
ONCOLOGY BREAKTHROUGHS
LUNG CANCER
SEVABERTINIB IN ADVANCED HER2-MUTANT NSCLC: A NEW ORAL TKI OPTION ACROSS TREATMENT LINES, INCLUDING POST-ADC DISEASE
BY ANNE JÄKEL
HER2 (ERBB2) activating mutations define a small but clinically important subset of non–small-cell lung cancer (NSCLC), affecting roughly 2–4% of patients and enriched in never-smokers and adenocarcinoma histology.1 Despite the broader success of precision oncology in NSCLC, HER2-mutant disease has historically lagged behind EGFR- and ALK-driven tumors, in part because early pan-HER tyrosine kinase inhibitors (TKIs) delivered modest efficacy at the cost of substantial toxicity.
The therapeutic landscape shifted with HER2-directed antibody–drug conjugates (ADCs), most notably trastuzumab deruxtecan (T-DXd), but interstitial lung disease (ILD) remains a pivotal safety consideration and post-ADC sequencing is an emerging challenge. In this context, the SOHO-01 trial positions sevabertinib (BAY 2927088), an oral, reversible, wild-type EGFR-sparing TKI designed to target mutant EGFR and HER2 (including exon 20 insertions), as a potentially versatile option across multiple clinical settings.1
Study design and patient population
SOHO-01 was an open-label, multicenter, international, multicohort phase 1–2 study evaluating sevabertinib at 20 mg twice daily in patients with locally advanced or metastatic HER2-mutant NSCLC. Efficacy was assessed by blinded independent central review, with objective response rate (ORR) as the primary endpoint in the expansion/extension cohorts and duration of response (DoR) and progression-free survival (PFS) as key secondary outcomes.
Patients were allocated into clinically pragmatic cohorts based on prior therapy: cohort D included previously treated patients without prior HER2- targeted therapy; cohort E comprised patients previously treated with HER2-directed ADCs; and cohort F enrolled treatment-naïve patients in the advanced/metastatic setting.1
As of the June 27, 2025 data cutoff, 209 patients had received sevabertinib (81 in cohort D, 55 in cohort E, 73 in cohort F). Follow-up was necessarily shorter in the first-line cohort (median 9.9 months in cohort F) than in previously treated cohorts (13.8 months in D; 11.7 months in E), reflecting later enrollment and ongoing treatment in a substantial proportion of untreated patients at analysis.
Baseline molecular features were consistent with known epidemiology: the majority of patients carried HER2 tyrosine kinase domain (TKD) mutations, and the exon 20 insertion Y772_A775dupYVMA was the dominant alteration, particularly in cohort F.1
Efficacy results
SOHO-01 reported robust antitumor activity in patients without prior HER2-targeted ADC exposure, with meaningful activity even after ADC therapy—an increasingly relevant population as ADC use expands.
Previously treated, HER2-therapy–naïve (cohort D): ORR was 64% (95% CI, 53–75). Median DoR was 9.2 months (95% CI, 6.3–13.5), and median PFS was 8.3 months (95% CI, 6.9–12.3).
Importantly, responses were also seen in heavily pretreated patients (including those with ≥2 prior lines), supporting sevabertinib’s activity beyond early-line use.1
Post-HER2 ADC (cohort E): ORR was 38% (95% CI, 25–52), with median DoR 8.5 months and median PFS 5.5 months. Among patients previously treated with T-DXd, approximately one-third responded, suggesting incomplete cross-resistance between ADC payload–mediated cytotoxicity and HER2 kinase inhibition.1
Treatment-naïve (cohort F): ORR was 71% (95% CI, 59–81), and median DoR was 11.0 months; PFS data were immature at cutoff. While cross-trial comparisons should be cautious, a ~70% response rate places sevabertinib in the emerging class of newer, HER2-selective (or mutant-selective) TKIs whose outcomes begin to approximate the response magnitude routinely expected in EGFR-driven NSCLC.1
Beyond conventional radiographic endpoints, SOHO-01 included exploratory signals relevant to clinical decision-making.
Responses were observed in patients with baseline brain metastases across cohorts, and the initial site of progression was the central nervous system (CNS) in only a small fraction of patients without baseline brain metastases, suggesting potential, but not definitive, CNS control.
These observations are hypothesis-generating given cohort size and the single-arm design.1
Safety profile
Tolerability is central for any HER2- targeted TKI in lung cancer, given the historical experience with pan-ERBB inhibitors and the clinical imperative to maintain dose intensity without excessive supportive-care burden. Across cohorts, grade ≥3 drug-related adverse events occurred in 31% of patients overall (range roughly 23–38% across cohorts), and diarrhea was the dominant toxicity, reported in 84–91% of patients, with grade ≥3 diarrhea occurring in 5–23% depending on cohort. Diarrhea led to dose reductions in a subset of patients but did not commonly drive discontinuation.
Overall, treatment was discontinued owing to drug-related adverse events in approximately 3% of patients, which presents an important signal that toxicity was generally manageable with contemporary dose-modification algorithms.1
A particularly notable finding was the absence of reported ILD/pneumonitis in this dataset. While vigilance remains warranted, especially as exposure time increases and use broadens into routine practice, this contrasts with the well-recognized ILD risk that accompanies T-DXd, which carries a boxed warning in the U.S. prescribing information.2
Clinical context
The key contribution of SOHO-01 is not simply that sevabertinib is active, but that it appears positionable across a modern HER2-mutant NSCLC treatment algorithm, including post-ADC disease, an area where evidence has been sparse and urgently needed.
T-DXd established HER2 as a clinically actionable driver in NSCLC and remains a major option for previously treated patients, but its ILD risk profile complicates patient selection and monitoring, and progression after ADC therapy is now an increasingly common clinical scenario.2
The emergence of oral HER2 TKIs expands the therapeutic toolkit and offers practical advantages: outpatient oral dosing, potential CNS penetration (still under study), and a mechanistically distinct approach that can be sequenced before or after ADCs.
SOHO-01 also lands in a rapidly evolving competitive space. The U.S. FDA granted accelerated approval to zongertinib in August 2025 for adults with unresectable or metastatic non-squamous NSCLC harboring HER2 TKD activating mutations after prior systemic therapy, formalizing the role of selective HER2 TKIs in routine care.3
The sevabertinib dataset provides complementary evidence supporting the broader concept: mutant-selective HER2 inhibition can deliver high response rates with acceptable tolerability, including in patients previously exposed to HER2 ADCs.
Finally, the exploratory biomarker work in cohort D adds a forward-looking layer.
Outcomes appeared more favorable in TKD mutations than non-TKD alterations, and ctDNA dynamics (clearance vs persistent detection early on treatment) separated PFS trajectories, suggesting a potential future role for ctDNA-guided risk stratification and adaptive treatment intensification.
These signals may not be practice-changing on their own, but they align with a broader movement toward integrating molecular response monitoring into targeted therapy trials.1
Conclusion
SOHO-01 positions sevabertinib as a clinically meaningful addition to the HER2-mutant NSCLC armamentarium, demonstrating high response rates in treatment-naïve and previously treated HER2-therapy–naïve patients, with sustained responses and measurable activity even after HER2-directed ADCs. Toxicity was dominated by diarrhea, with relatively low discontinuation rates and no reported ILD/pneumonitis in this analysis.1
As HER2-mutant NSCLC care becomes increasingly pathway- and sequence-driven, sevabertinib underscores the momentum behind mutant-selective HER2 TKIs, and sets the stage for randomized confirmation and clearer positioning relative to ADCs and other targeted agents.
Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029-33.
Liberti MV, Locasale JW. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem Sci. 2016;41(3):211-8.
Danzi F, Pacchiana R, Mafficini A, Scupoli MT, Scarpa A, Donadelli M, et al. To metabolomics and beyond: a technological portfolio to investigate cancer metabolism. Signal Transduct Target Ther. 2023;8(1):137.
Keoh LQ, Chiu CF, Ramasamy TS. Metabolic Plasticity and Cancer Stem Cell Metabolism: Exploring the Glycolysis-OXPHOS Switch as a Mechanism for Resistance and Tumorigenesis. Stem Cell Rev Rep. 2025;21(8):2446-68.
Al-Horani RA. Cancer Metabolism and Its Historical & Molecular Foundations: An Overview. Drugs and Drug Candidates. 2026;5(1).
Ramanujan VK. Metabolic Plasticity in Cancer Cells: Reconnecting Mitochondrial Function to Cancer Control. J Cell Sci Ther. 2015;6(3).
Nong S, Han X, Xiang Y, Qian Y, Wei Y, Zhang T, et al. Metabolic reprogramming in cancer: Mechanisms and therapeutics. MedComm (2020). 2023;4(2):e218.
Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer. 2016;16(10):619-34.
Jin J, Byun JK, Choi YK, Park KG. Targeting glutamine metabolism as a therapeutic strategy for cancer. Exp Mol Med. 2023;55(4):706-15.
Broadfield LA, Pane AA, Talebi A, Swinnen JV, Fendt SM. Lipid metabolism in cancer: New perspectives and emerging mechanisms. Dev Cell. 2021;56(10):1363-93.
Jonker PB, Muir A. Metabolic ripple effects - deciphering how lipid metabolism in cancer interfaces with the tumor microenvironment. Dis Model Mech. 2024;17(9).
Chen J, Huang Z, Chen Y, Tian H, Chai P, Shen Y, et al. Lactate and lactylation in cancer. Signal Transduct Target Ther. 2025;10(1):38.
Faubert B, Li KY, Cai L, Hensley CT, Kim J, Zacharias LG, et al. Lactate Metabolism in Human Lung Tumors. Cell. 2017;171(2):358-71 e9.
de Visser KE, Joyce JA. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell. 2023;41(3):374-403.
Ahmed MAM, Nagelkerke A. Current developments in modelling the tumour microenvironment in vitro: Incorporation of biochemical and physical gradients. Organs-on-a-Chip. 2021;3.
Chai X, Tao Q, Li L. Spatiotemporal Heterogeneity of Tumor Glucose Metabolism Reprogramming: From Single-Cell Mechanisms to Precision Interventions. Int J Mol Sci. 2025;26(14).
Kim EY, Abides J, Keller CR, Martinez SR, Li W. Tumor Microenvironment Lactate: Is It a Cancer Progression Marker, Immunosuppressant, and Therapeutic Target? Molecules. 2025;30(8).
Cassim S, Pouyssegur J. Tumor Microenvironment: A Metabolic Player that Shapes the Immune Response. Int J Mol Sci. 2019;21(1).
Li S, Zhang Y, Tong H, Sun H, Liao H, Li Q, et al. Metabolic regulation of immunity in the tumor microenvironment. Cell Rep. 2025;44(11):116463.
Christensen DS, Ahrenfeldt J, Sokac M, Kisistok J, Thomsen MK, Maretty L, et al. Treatment Represents a Key Driver of Metastatic Cancer Evolution. Cancer Res. 2022;82(16):2918-27.
Zhu Y, Yan W, Tong L, Yang J, Ge S, Fan J, et al. Metabolic Reprogramming: A Crucial Contributor to Anticancer Drug Resistance. MedComm (2020). 2025;6(9):e70358.
Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sanchez N, Marchesini M, et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature. 2014;514(7524):628-32.
DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475(7354):106-9.
Khan T, Nagarajan M, Kang I, Wu C, Wangpaichitr M. Targeting Metabolic Vulnerabilities to Combat Drug Resistance in Cancer Therapy. J Pers Med. 2025;15(2).
Kang YP, Ward NP, DeNicola GM. Recent advances in cancer metabolism: a technological perspective. Exp Mol Med. 2018;50(4):1-16.
Guerrero-Lopez P, Martin-Pardillos A, Bonet-Aleta J, Mosseri A, Hueso JL, Santamaria J, et al. 2D versus 3D tumor-on-chip models to study the impact of tumor organization on metabolic patterns in vitro. Sci Rep. 2025;15(1):19506.
Jang C, Chen L, Rabinowitz JD. Metabolomics and Isotope Tracing. Cell. 2018;173(4):822-37.
Reinfeld BI, Madden MZ, Wolf MM, Chytil A, Bader JE, Patterson AR, et al. Cell-programmed nutrient partitioning in the tumour microenvironment. Nature. 2021;593(7858):282-8.
Faubert B, Solmonson A, DeBerardinis RJ. Metabolic reprogramming and cancer progression. Science. 2020;368(6487).
Luengo A, Gui DY, Vander Heiden MG. Targeting Metabolism for Cancer Therapy. Cell Chem Biol. 2017;24(9):1161-80.
Martinez-Reyes I, Chandel NS. Cancer metabolism: looking forward. Nat Rev Cancer. 2021;21(10):669-80
LOBBYING FOR CHANGE
WHEN YOUTH DOESN’T EQUATE SAFETY
CoppaFeel! and the cost of being “too young” for breast cancer
Partridge AH, Hughes ME, Warner ET, Ottesen RA, Wong YN, Edge SB, Theriault RL, Blayney DW, Niland JC, Winer EP, Weeks JC. Subtype-dependent relationship between young age at diagnosis and breast cancer survival. Journal of Clinical Oncology. 2016 Sep 20;34(27):3308-14.
Anders CK, Johnson R, Litton J, Phillips M, Bleyer A. Breast cancer before age 40 years. InSeminars in oncology 2009 Jun 1 (Vol. 36, No. 3, pp. 237-249). WB Saunders.
Ramirez AJ, Westcombe AM, Burgess CC, Sutton S, Littlejohns P, Richards MA. Factors predicting delayed presentation of symptomatic breast cancer: a systematic review. The Lancet. 1999 Apr 3;353(9159):1127-31.
Ruddy KJ, Gelber S, Tamimi RM, Schapira L, Come SE, Meyer ME, Winer EP, Partridge AH. Breast cancer presentation and diagnostic delays in young women. Cancer. 2014 Jan 1;120(1):20-5.
Arndt V, Stürmer T, Stegmaier C, Ziegler H, Dhom G, Brenner H. Patient delay and stage of diagnosis among breast cancer patients in Germany–a population based study. British journal of cancer. 2002 Apr;86(7):1034-40.
Richards MA, Westcombe AM, Love SB, Littlejohns P, Ramirez AJ. Influence of delay on survival in patients with breast cancer: a systematic review. The Lancet. 1999 Apr 3;353(9159):1119-26.
Heer E, Harper A, Escandor N, Sung H, McCormack V, Fidler-Benaoudia MM. Global burden and trends in premenopausal and postmenopausal breast cancer: a population-based study. The Lancet Global Health. 2020 Aug 1;8(8):e1027-37.
Partridge AH, Hughes ME, Warner ET, Ottesen RA, Wong YN, Edge SB, Theriault RL, Blayney DW, Niland JC, Winer EP, Weeks JC. Subtype-dependent relationship between young age at diagnosis and breast cancer survival. Journal of Clinical Oncology. 2016 Sep 20;34(27):3308-14.
Anders CK, Hsu DS, Broadwater G, Acharya CR, Foekens JA, Zhang Y, Wang Y, Marcom PK, Marks JR, Febbo PG, Nevins JR. Young age at diagnosis correlates with worse prognosis and defines a subset of breast cancers with shared patterns of gene expression. Journal of clinical oncology. 2008 Jul 10;26(20):3324-30.
Azim HA, Partridge AH. Biology of breast cancer in young women. Breast cancer research. 2014 Aug 27;16(4):427.
CIRCULAR DNA DRIVERS
Extrachromosomal DNA: a hidden engine of cancer evolution
Turner KM, Deshpande V, Beyter D, Koga T, Rusert J, Lee C, et al. Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature. 2017;543(7643):122-5.
Storlazzi CT, Lonoce A, Guastadisegni MC, Trombetta D, D’Addabbo P, Daniele G, et al. Gene amplification as double minutes or homogeneously staining regions in solid tumors: origin and structure. Genome Res. 2010;20(9):1198-206.
Bailey C, Pich O, Thol K, Watkins TBK, Luebeck J, Rowan A, et al. Origins and impact of extrachromosomal DNA. Nature. 2024;635(8037):193-200.
Bafna V, Mischel PS. Extrachromosomal DNA in Cancer. Annu Rev Genomics Hum Genet. 2022;23:29-52.
Chapman OS, Luebeck J, Sridhar S, Wong IT, Dixit D, Wang S, et al. Circular extrachromosomal DNA promotes tumor heterogeneity in high-risk medulloblastoma. Nat Genet. 2023;55(12):2189-99.
Ma X, Yu X, Wu C, Song L. The role of extrachromosomal DNA in tumorigenesis and progression. Front Oncol. 2025;15:1665024.
Yan X, Mischel P, Chang H. Extrachromosomal DNA in cancer. Nat Rev Cancer. 2024;24(4):261-73.
Wu Y, Rui R, Tian T, Zheng J, He S, Zhou L, et al. Extrachromosomal DNA and cancer: function, formation, and clinical implications. J Exp Clin Cancer Res. 2026;45(1):50.
`Yang QL, Xie Y, Qiao K, Lim JYS, Wu S. Modern biology of extrachromosomal DNA: A decade-long voyage of discovery. Cell Res. 2025;35(1):11-22.
Kim H, Nguyen NP, Turner K, Wu S, Gujar AD, Luebeck J, et al. Extrachromosomal DNA is associated with oncogene amplification and poor outcome across multiple cancers. Nat Genet. 2020;52(9):891-7.
Lange JT, Rose JC, Chen CY, Pichugin Y, Xie L, Tang J, et al. The evolutionary dynamics of extrachromosomal DNA in human cancers. Nat Genet. 2022;54(10):1527-33.
Dong Y, He Q, Chen X, Yang F, He L, Zheng Y. Extrachromosomal DNA (ecDNA) in cancer: mechanisms, functions, and clinical implications. Front Oncol. 2023;13:1194405.
Wu X, Li P, Yimiti M, Ye Z, Fang X, Chen P, et al. Identification and Characterization of Extrachromosomal Circular DNA in Plasma of Lung Adenocarcinoma Patients. Int J Gen Med. 2022;15:4781-91.
Chapman OS, Sridhar S, Chow EY, Kenkre R, Kirkland J, Dutta A, et al. Extrachromosomal DNA associates with poor survival across a broad spectrum of childhood solid tumors. medRxiv. 2025.
Khatami F, Larijani B, Tavangar SM. The presence of tumor extrachomosomal circular DNA (ecDNA) as a component of liquid biopsy in blood. Med Hypotheses. 2018;114:5-7.
Zhu X, Qian Y, Tang Q, Li J, Geng Y, Gong S, et al. Research Advances of Extrachromosomal Circular DNA in Breast Cancer. Cancer Med. 2025;14(19):e71285.
Nathanson DA, Gini B, Mottahedeh J, Visnyei K, Koga T, Gomez G, et al. Targeted therapy resistance mediated by dynamic regulation of extrachromosomal mutant EGFR DNA. Science. 2014;343(6166):72-6.
Yuan XQ, Zhou N, Song SJ, Xie YX, Chen SQ, Yang TF, et al. Decoding the genomic enigma: Approaches to studying extrachromosomal circular DNA. Heliyon. 2024;10(17):e36659.
Zhang Y, Chen R, Gan H. Unfolding ecDNA as a pan-cancer therapeutic target. Med Rev (2021). 2025;5(5):432-5.
Wong IT, Yi H, Melillo B, Cravatt BF, Chang HY, Mischel PS. Targeting extrachromosomal DNA in human cancers. Nat Rev Drug Discov. 2026.
ONCOLOGY BREAKTHROUGHS
MULTIPLE MYELOMA
IFM2017-03: dexamethasone-sparing daratumumab–lenalidomide sets a new benchmark for frail, newly diagnosed myeloma
Manier S, Lambert J, Hulin C, Macro M, Laribi K, Araujo C, Pica GM, Touzeau C, Godmer P, Slama B, Karlin L. Safety and efficacy of a dexamethasone-sparing regimen with daratumumab and lenalidomide in patients with frailty and newly diagnosed multiple myeloma (IFM2017-03): a phase 3, open-label, multicentre, randomised, controlled trial. The Lancet Oncology. 2025 Oct 1;26(10):1323-33.
Facon T, Moreau P, Weisel K, Goldschmidt H, Usmani SZ, Chari A, Plesner T, Orlowski RZ, Bahlis N, Basu S, Hulin C. Daratumumab/lenalidomide/dexamethasone in transplant-ineligible newly diagnosed myeloma: MAIA long-term outcomes. Leukemia. 2025 Apr;39(4):942-50.
Palumbo A, Bringhen S, Mateos MV, Larocca A, Facon T, Kumar SK, Offidani M, McCarthy P, Evangelista A, Lonial S, Zweegman S. Geriatric assessment predicts survival and toxicities in elderly myeloma patients: an International Myeloma Working Group report. Blood, The Journal of the American Society of Hematology. 2015 Mar 26;125(13):2068-74.
LUNG CANCER
Sevabertinib in advanced HER2-mutant NSCLC: a new oral TKI option across treatment lines, including post-ADC disease
Le X, Kim TM, Loong HH, Prelaj A, Goh BC, Li L, Fang Y, Lu S, Dong X, Wu L, Shinno Y. Sevabertinib in advanced HER2-mutant non–small-cell lung cancer. New England Journal of Medicine. 2025 Nov 6;393(18):1819-32.
Oncology Compass Digest presents a selection of medical conferences happening this spring. The Oncology Compass Calendar is the most comprehensive calendar of global oncology conferences.
ONCOLOGY COMPASS IS GLOBALLY BECOMING AN INCREASINGLY IMPORTANT PLATFORM FOR ONCOLOGISTS
17 Scientific Leaders
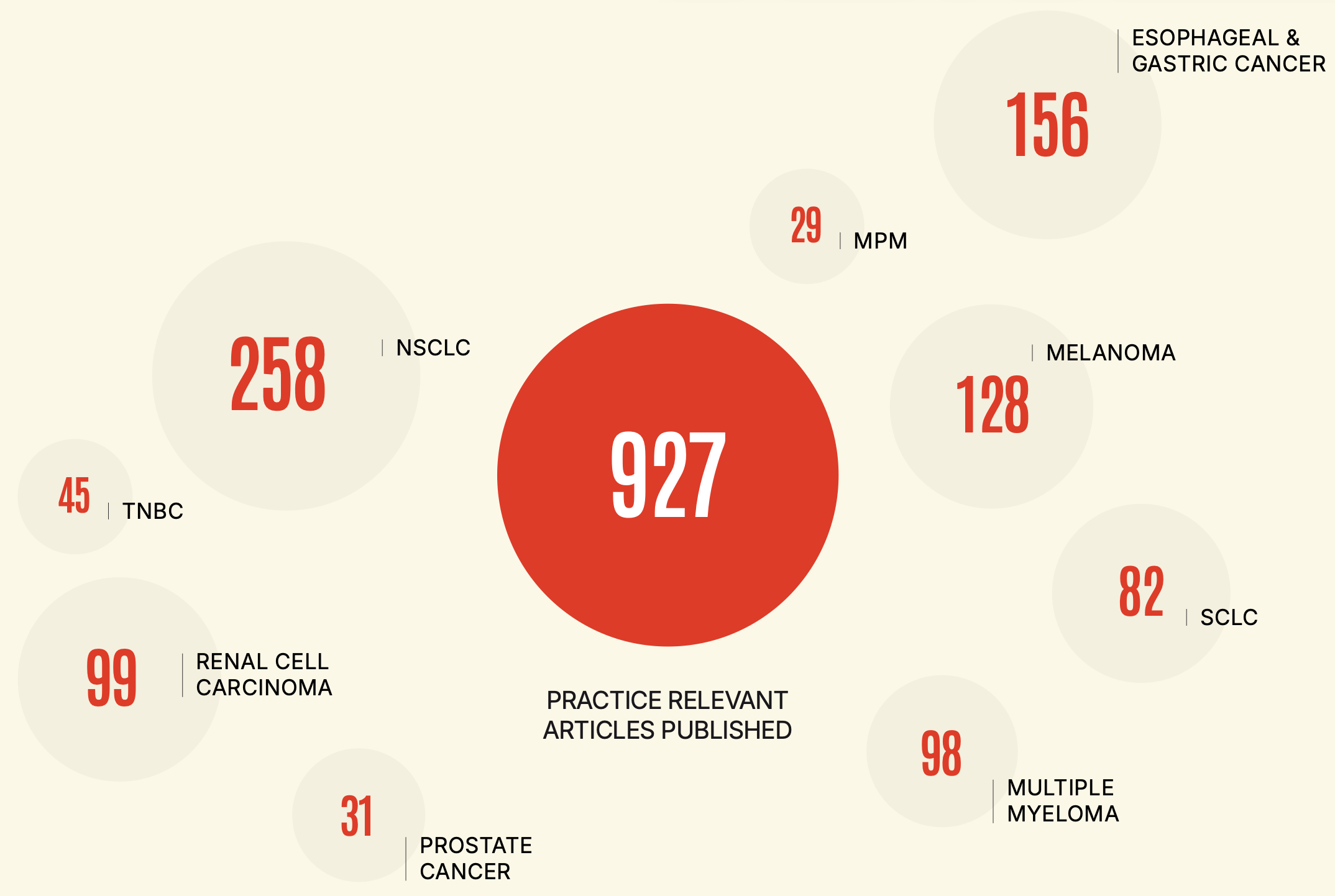
selected practice-relevant publications for lung, renal, gastro-esophageal, melanoma, multiple myeloma, prostate cancer and triple-negative breast cancer
505
ACTIVE users
TOP 3 FILTER CRITERIA:
MELANOMA
294
ESOPHAGEAL & GASTRIC
295
NSCLC
419
*Total number of clicks on filter criteria over time
Website Visitors
7,950
Pageviews
12,850
Sessions
8,360
Avg. Session
05:10
Pages per session
1.54
Avg. Session Duration per Returning User
6:30 min
Pages per Session per Returning User
5.14
USER DATA & WEBSITE INSIGHTS
VISITORS by DEVICES
device category
total visitors
pageviews
Mobile
4,284
7,132
Desktop
3,695
5,877
Tablet
213
300
TOP 10 COUNTRIES WHERE VISITORS COME FROM:
country
VISITORS
PAGEVIEWS
1. United States
1,467
1,523
2. China
1,127
1,130
3. Ireland
1,040
1,162
4. Romania
477
502
5. United Kingdom
475
492
6. Spain
376
414
7. Singapore
336
336
8. Malta
277
344
9. Switzerland
215
238
10. Sweden
150
158
The number of Visitors represents all visitors to Oncology Compass, both registered and non-registered users. The metrics for Users relate to the Registered Users data who have full access to the Oncology Compass platform.
VISITORS BY AGE / GENDER*
age
VISITORS
pageviews
1. 65+
615
701
2. 45-54
350
390
3. 55-64
329
371
4. 25-34
299
316
5. 35-44
208
227
6. 18-24
90
95
VISITORS BY GENDER *
60.9%
Total female
39.1%
Total female
* for the rest of visitors, the gender and age are unknown, they choose not to share it.
Oncology Compass Digest is edited and hosted by Capptoo AG, incorporated in Zürich, Switzerland.
Information related to any product(s) may not be consistent with the prescribing information. The Digest editorial team has made a constant care to make sure that content is accurate on the date of publication. The views expressed in the articles reflect the opinions of the interviewed persons and are not necessarily the views of the publisher. All rights reserved. No content can be partially or wholly reprinted or reproduced.
Published by: Capptoo AG, Churerstrasse 92i, 8808 Pfäffikon SZ, Switzerland. Any text and design are the property of Capptoo AG. Any other use of the materials is prohibited unless Capptoo has previously agreed.